Find all inteins in Archaea genomes
Pariksheet Nanda
2018-05-02
intein-finder.RmdBackground
Project goal
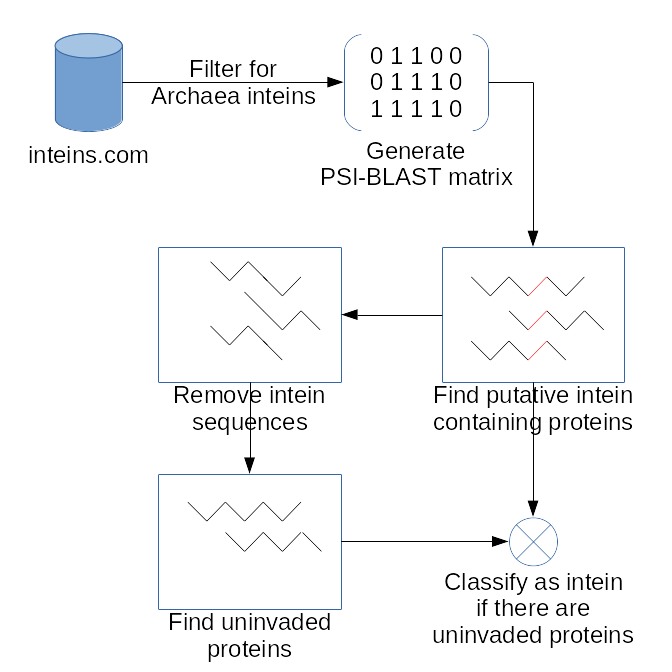
The project goal is to identify new inteins in silico. In general the approach is to:
- Find distant intein homologs using PSI-BLAST.
- Remove the intein sequences from the homologs, and query BLASTP to verify that there are versions of the protein not invaded by the intein.
The is illustrated in slightly more detail in the workflow below:

Further work
Focus on identifying splicing domains (A, B, F, G) and homing endonucleases (C, D, E, H); not necessarily all the annotated domains which have lots of variation.
Then do a phylogeny of the homing endonucleases and see if they are really vertically inherited or see if they horizontally jump back and forth and are the basis of some phylogenies.
Find putative intein containing proteins
Fetch InBase Archaea
Extract Archaea intein sequences
suppressPackageStartupMessages({
library(tibble) # Load data as tibble instead of data.frame
library(inbaser)
library(Biostrings) # readAAStringSet, writeAAStringSet
library(dplyr) # %>%
library(tidyr) # gather
library(stringr) # str_remove
})
data(inbase)
## Add row numbers to make it possible to subset ibase_seq.
inbase <- inbase %>%
mutate(i = row_number())
## Motifs.
all_endo <- c("C", "D", "E", "H")
## Having all 4 splicing domains is criterion #3 per inbase.com intein
## designation.
blocks <-
inbase %>%
## Gather also removes the NA values.
gather("Block", "Sequence", starts_with("Block")) %>%
select(Sequence, everything()) %>%
## Set "None" values to NA
mutate(Sequence = ifelse(str_detect(Sequence, "(None|[-]+)"),
NA, Sequence)) %>%
mutate(Block = str_remove(Block, "Block ")) %>%
mutate(has_endo = Block %in% all_endo) %>%
select(i, Block, Sequence, has_endo, everything()) %>%
arrange(i) %>%
group_by(i) %>%
summarize(has_endo = sum(! is.na(Sequence) & has_endo)) %>%
filter(has_endo == max(has_endo))
## Subset to archaea.
archaea <- inbase %>%
inner_join(blocks, by = "i") %>%
filter(`Domain of Life` == "Archaea")
## Get corresponding sequences.
seq <- inbase_seq[archaea %>% pull(i)]Export the protein sequences and run the alignment on the cluster in psi-blast.sh:
writeXStringSet(seq, "archaea_inteins.faa")Train PSI-blast matrix using NR
The NR database on the BBC server is located in /common/blast/data/. Note the -num_iterations 0 feature of psiblast that auto-calculates the matrix optimal iteration value.
Script psiblast-train.sh to train PSI-BLAST weight matrix:
#!/bin/bash
# Blast against non-redundant database
prefix_database=/common/blast/data/nr
# Only Blast against these all Archaea proteins in the database
file_subject=archaea.gi
# Archaea inteins
file_query_raw=archaea_inteins.faa
file_query=${file_query_raw%.faa}.msa
if ! [[ -f $file_query ]]; then
muscle -in $file_query_raw -out $file_query
fi
psiblast \
-num_threads `nproc` \
-num_iterations 0 \
-in_msa $file_query \
-db $prefix_database \
-gilist $file_subject \
-outfmt 7 \
-out out.blast-train \
-out_pssm out.pssmThe search converges with a 1.25 hour wall time and our position matrix is saved to out.pssm.
Job output in out.qsub-train:
Job ID : 790294
Date : Wed Apr 18 15:08:00 EDT 2018
Script : /opt/gridengine/default/spool/compute-1-7/job_scripts/790294
Queue : course.q
Par Env :
psiblast: /lib64/libz.so.1: no version information available (required by psiblast)
Warning: [psiblast] lcl|1: Warning: Composition-based score adjustment conditioned on sequence properties and unconditional composition-based score adjustment is not supported with PSSMs, resetting to default value of standard composition-based statistics
Command being timed: "bash psiblast-train.sh"
User time (seconds): 16542.95
System time (seconds): 243.25
Percent of CPU this job got: 368%
Elapsed (wall clock) time (h:mm:ss or m:ss): 1:15:52
Average shared text size (kbytes): 0
Average unshared data size (kbytes): 0
Average stack size (kbytes): 0
Average total size (kbytes): 0
Maximum resident set size (kbytes): 3816192
Average resident set size (kbytes): 0
Major (requiring I/O) page faults: 2971
Minor (reclaiming a frame) page faults: 116847626
Voluntary context switches: 1144313
Involuntary context switches: 108241
Swaps: 0
File system inputs: 71666856
File system outputs: 339064
Socket messages sent: 0
Socket messages received: 0
Signals delivered: 0
Page size (bytes): 4096
Exit status: 0Job submission script train.qsub:
#!/usr/bin/env bash
#$ -N psiblast_train
#$ -q course.q
#$ -o out.qsub-train
#$ -j yes
#$ -S /bin/bash
#$ -cwd
#$ -M NAME.REDACTED@uconn.edu
#$ -m ea
# ^ Directives for Sun Grid Engine.
# Clear output file.
echo -n > out.qsub-train
# Job information.
echo "
Job ID=: $JOB_ID
Date=: $(date)
Script=: $JOB_SCRIPT
Queue=: $QUEUE
Par Env=: $PE
" | column -t -s '='
# Load psiblast software module.
module purge
module load blast/2.6.0
# Run psiblast.
command time -v bash psiblast-train.shRun PSI-blast against NR
Script psiblast-run.sh uses PSI-BLAST weight matrix to return proteins containing inteins:
#!/bin/bash
# Blast against non-redundant database
prefix_database=/common/blast/data/nr
# Only Blast against these all Archaea proteins in the database
file_subject=archaea.gi
psiblast \
-num_threads `nproc` \
-in_pssm out.pssm \
-db $prefix_database \
-gilist $file_subject \
-outfmt 7 \
-out out.blast-runThe run only takes a minute per out.qsub-run:
Job ID : 790767
Date : Mon Apr 30 12:55:14 EDT 2018
Script : /opt/gridengine/default/spool/compute-1-9/job_scripts/790767
Queue : course.q
Par Env :
psiblast: /lib64/libz.so.1: no version information available (required by psiblast)
Warning: [psiblast] lcl|1: Warning: Composition-based score adjustment conditioned on sequence properties and unconditional composition-based score adjustment is not supported with PSSMs, resetting to default value of standard composition-based statistics
Command being timed: "bash psiblast-run.sh"
User time (seconds): 62.29
System time (seconds): 16.20
Percent of CPU this job got: 20%
Elapsed (wall clock) time (h:mm:ss or m:ss): 6:30.30
Average shared text size (kbytes): 0
Average unshared data size (kbytes): 0
Average stack size (kbytes): 0
Average total size (kbytes): 0
Maximum resident set size (kbytes): 2578592
Average resident set size (kbytes): 0
Major (requiring I/O) page faults: 2835
Minor (reclaiming a frame) page faults: 670392
Voluntary context switches: 88741
Involuntary context switches: 108333
Swaps: 0
File system inputs: 65933576
File system outputs: 144
Socket messages sent: 0
Socket messages received: 0
Signals delivered: 0
Page size (bytes): 4096
Exit status: 0Find corresponding uninvaded proteins
Read in PSI-BLAST results table
library(readr)
library(stringr)
library(DT) # datatable
read_blast <- function(file, ...) {
header <- read_lines("out.blast-run", skip = 4, n_max = 1) %>%
str_remove("^# Fields: ") %>%
str_split(", ", simplify = TRUE) %>%
str_replace("%", "percent") %>%
str_replace_all("[ .]+", "_")
read_delim(file, "\t", col_names = header, comment = "#", ...)
}
tbl <- read_blast("out.blast-run")%>%
select(-query_acc_ver)
#> Parsed with column specification:
#> cols(
#> query_acc_ver = col_integer(),
#> subject_acc_ver = col_character(),
#> percent_identity = col_double(),
#> alignment_length = col_integer(),
#> mismatches = col_integer(),
#> gap_opens = col_integer(),
#> q_start = col_integer(),
#> q_end = col_integer(),
#> s_start = col_integer(),
#> s_end = col_integer(),
#> evalue = col_double(),
#> bit_score = col_double()
#> )
## Print paginated table.
datatable(tbl)Find uninvaded proteins which do not have the intein
Now that we have proteins with the intein, find a corresponding protein sequence with the intein removed. Remove the intein and run BLAST on the removed sequences.
We can save our list of accessions to query from the database.
tbl %>%
pull(subject_acc_ver) %>%
## By default, psiblast returns max 500 results, and many of the
## subject results are duplicated.
unique() %>%
tibble::as.tibble() %>%
write_csv("inteins_putative.accessions", col_names = FALSE)Query the sequences from the database using blastdbcmd as shown in this file blastdbcmd-sequences.sh. This is quick and only takes a few seconds:
#!/bin/bash
# Blast against non-redundant database
prefix_database=/common/blast/data/nr
file_entry=inteins_putative.accessions
file_seq=${file_entry%.accessions}.faa
blastdbcmd \
-entry_batch $file_entry \
-db $prefix_database \
-out $file_seqRead the proteins back into R to remove the intein segments.
proteins_with_intein <- readAAStringSet("inteins_putative.faa")Discovered multiple hits for the same protein
library(ggplot2)
counts <-
tbl %>%
add_count(subject_acc_ver) %>%
select(n, everything()) %>%
arrange(desc(n))
ggplot(counts %>% group_by(subject_acc_ver) %>% top_n(1),
aes(x = n)) +
stat_bin(bins = max(counts$n)) +
stat_bin(aes(label = ..count..), geom = "text", vjust = -0.5,
bins = max(counts$n)) +
labs(title = sprintf("Duplication of hits (total = %d, unique = %d)", nrow(tbl), length(unique(tbl$subject_acc_ver))),
x = "duplicate hits")
#> Selecting by bit_score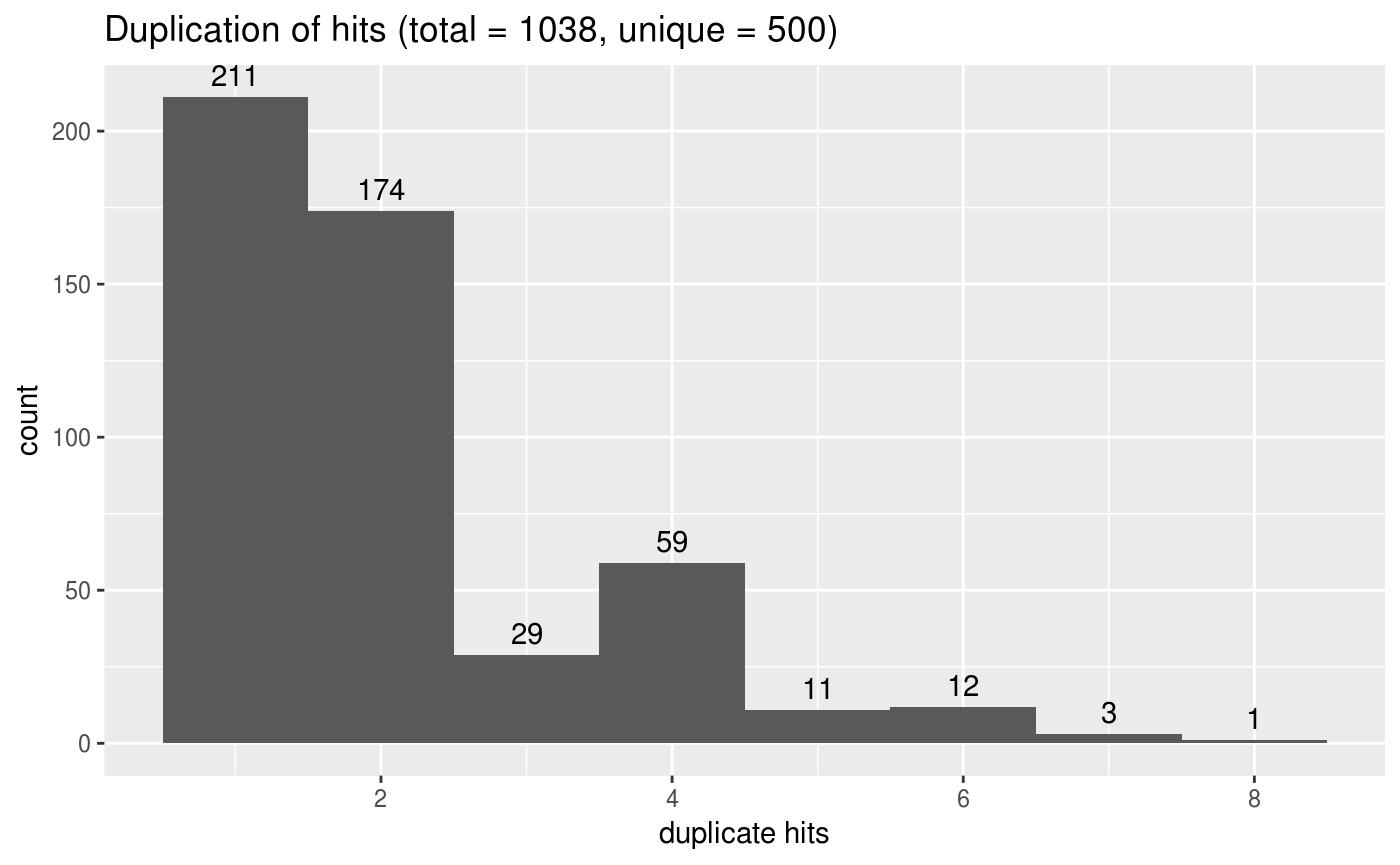
Dyanna suggested one strategy for removing duplicate results is removing results with low alignment length values:
alignment_length_cutoff <- width(seq) %>% min() * 0.9
ggplot(counts %>% mutate(keep = alignment_length >= alignment_length_cutoff),
aes(x = alignment_length, fill = keep)) +
geom_histogram(binwidth = 10) +
## FIXME: In future relax this 90% minimum length threshold and
## confirm the intein is in the
geom_vline(xintercept = alignment_length_cutoff) +
labs(title = "Using 80% minimum alignment length to remove extra hits",
x = "alignment length in nucleotides, not amino acids!",
fill = "keep hit?") +
facet_wrap(~ n) +
theme(legend.position = "top")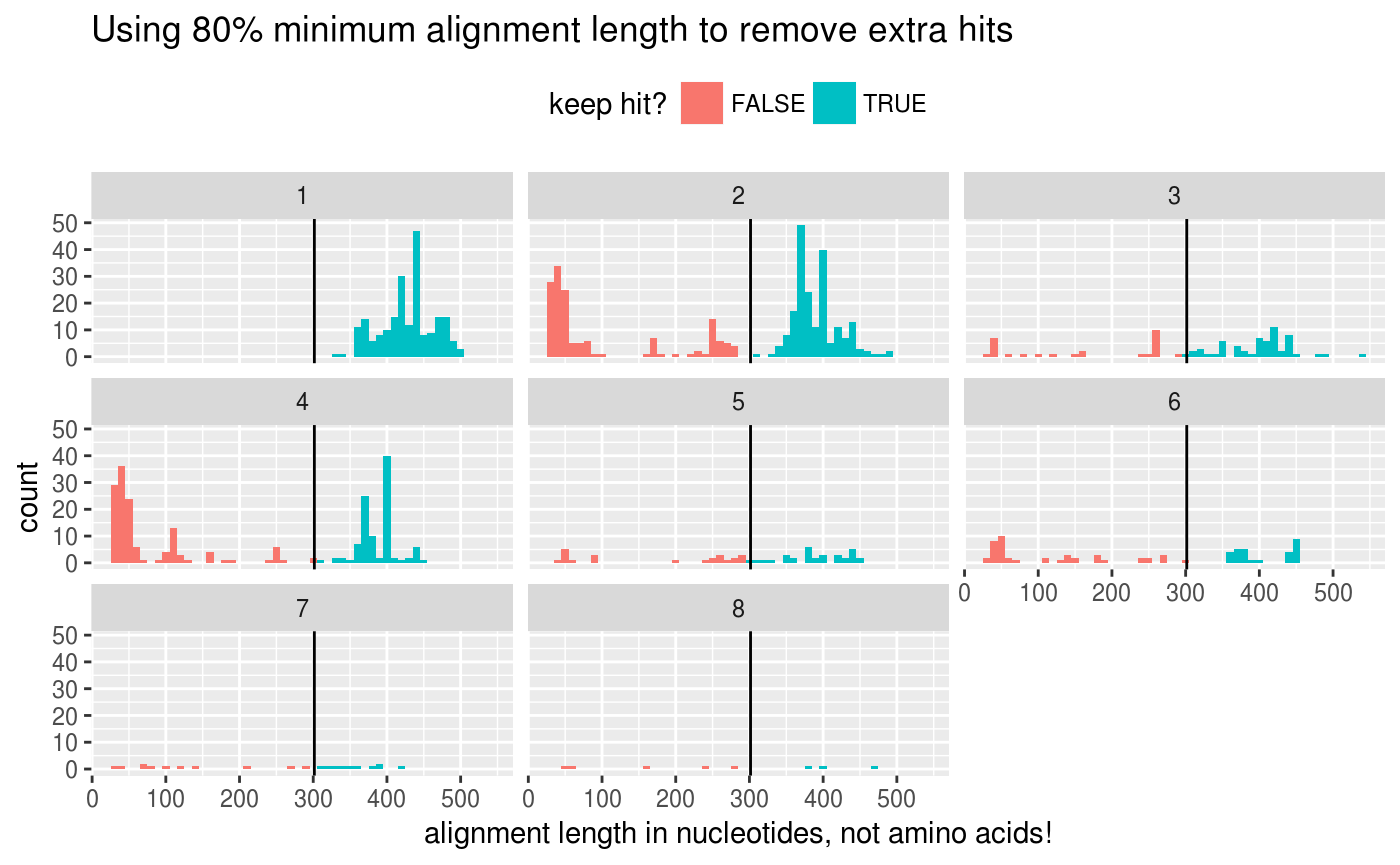
Filter hits by the alignment length:
Remove intein from protein hits
## The NR naming always puts the accession identifier as the first
## word and the description separated by a space. To be able to match
## up the names with our blast results, only keep the accession
## identifier and remove the description.
names(proteins_with_intein) <- names(proteins_with_intein) %>%
str_remove(" .+")
proteins_without_intein <- proteins_with_intein[hits %>% pull(subject_acc_ver)]
subseq(proteins_without_intein,
start = hits %>% pull(s_start),
end = hits %>% pull(s_end)) <- NULL
names(proteins_without_intein) <- paste0(names(proteins_without_intein),
"_delta_",
hits %>% pull(s_start),
"-",
hits %>% pull(s_end))
writeXStringSet(proteins_without_intein, "proteins_intein_free.faa")Blastp intein-free protein hits to find non-intein proteins
Use our intein-free protein to find whether we can get better results than the original protein.
#!/bin/bash
# Blast against non-redundant database
prefix_database=/common/blast/data/nr
# Blast all intein-free proteins
file_query=proteins_intein_free.faa
# Only Blast against these all Archaea proteins in the database
file_subject=archaea.gi
blastp \
-num_threads `nproc` \
-query $file_query \
-db $prefix_database \
-gilist $file_subject \
-outfmt 7 \
-out out.blast-intein-freehits_all <- read_blast("out.blast-intein-free.bz2")
#> Parsed with column specification:
#> cols(
#> query_acc_ver = col_character(),
#> subject_acc_ver = col_character(),
#> percent_identity = col_double(),
#> alignment_length = col_integer(),
#> mismatches = col_integer(),
#> gap_opens = col_integer(),
#> q_start = col_integer(),
#> q_end = col_integer(),
#> s_start = col_integer(),
#> s_end = col_integer(),
#> evalue = col_double(),
#> bit_score = col_double()
#> )
hits_top <- hits_all %>%
mutate(query_orig = str_remove(query_acc_ver, "_delta_.+")) %>%
select(query_orig, subject_acc_ver, everything()) %>%
## Drop self-hits.
filter(subject_acc_ver != query_orig) %>%
## Quality filter.
filter(evalue < 1e-5) %>%
## Top hits.
group_by(query_orig, subject_acc_ver) %>%
top_n(1, bit_score) %>%
add_count(query_orig)## The dataframe is quite large, so only show a small portion.
nrow(hits_top)
#> [1] 163415
datatable(hits_top %>% head(1e3))ggplot(hits_top, aes(n)) +
stat_bin(bins = max(hits_top$n)) +
stat_bin(aes(label = ..count..), geom = "text", vjust = -0.5,
bins = max(hits_top$n)) +
## Show all ticks, not just 2, 4, 6.
scale_x_continuous(breaks = 1:max(hits_top$n)) +
scale_y_log10()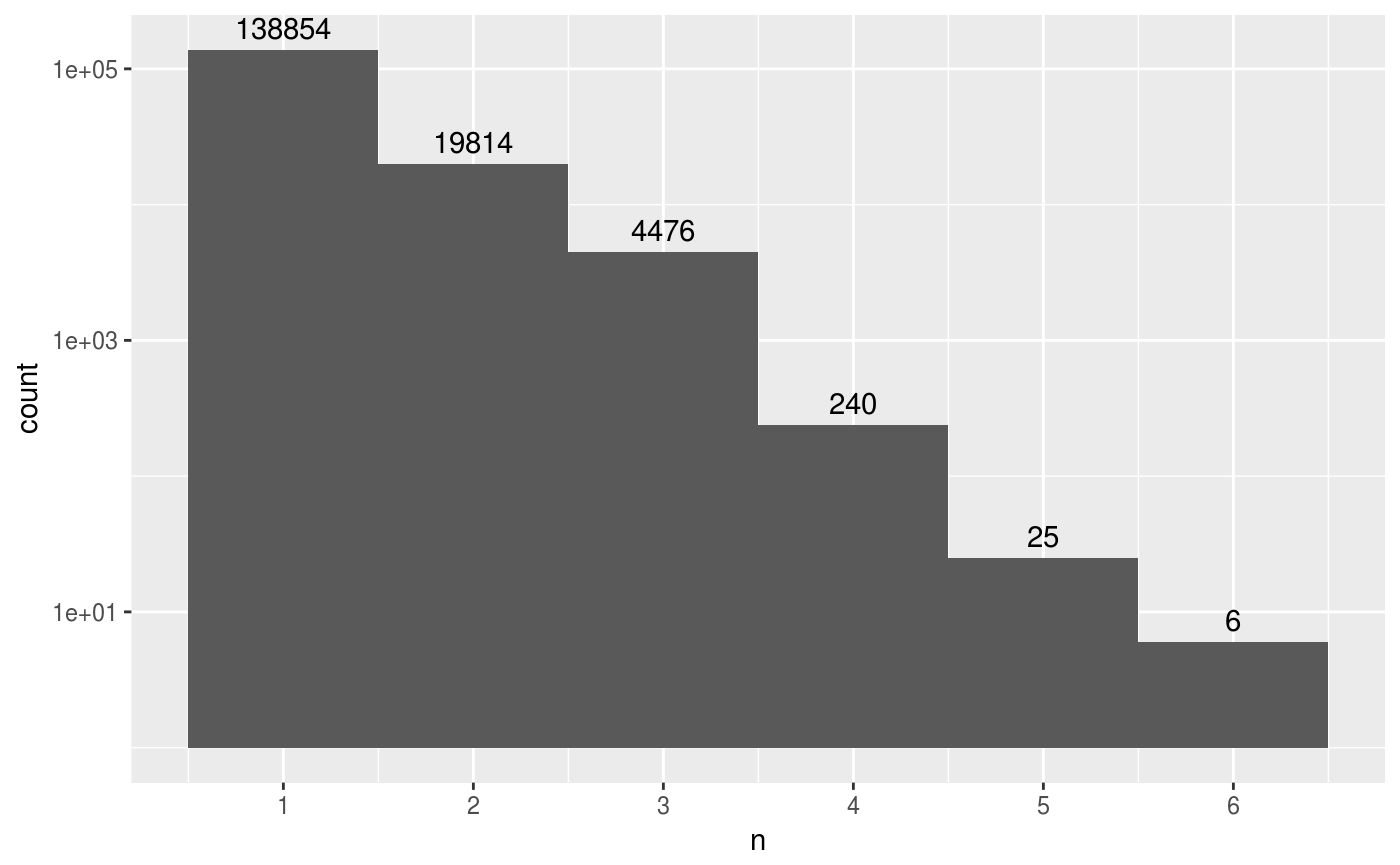
Session Info
sessionInfo()
#> R version 3.5.0 (2017-01-27)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 14.04.5 LTS
#>
#> Matrix products: default
#> BLAS: /home/travis/R-bin/lib/R/lib/libRblas.so
#> LAPACK: /home/travis/R-bin/lib/R/lib/libRlapack.so
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> attached base packages:
#> [1] stats4 parallel stats graphics grDevices utils datasets
#> [8] methods base
#>
#> other attached packages:
#> [1] ggplot2_2.2.1 DT_0.4.9 readr_1.1.1
#> [4] bindrcpp_0.2.2 stringr_1.3.0 tidyr_0.8.0
#> [7] dplyr_0.7.4 Biostrings_2.45.3 XVector_0.20.0
#> [10] IRanges_2.14.0 S4Vectors_0.18.0 BiocGenerics_0.26.0
#> [13] inbaser_0.0.0.9000 tibble_1.4.2
#>
#> loaded via a namespace (and not attached):
#> [1] tidyselect_0.2.4 purrr_0.2.4 colorspace_1.3-2 htmltools_0.3.6
#> [5] yaml_2.1.19 rlang_0.2.0 pkgdown_1.0.0 pillar_1.2.2
#> [9] later_0.7.2 glue_1.2.0 bindr_0.1.1 plyr_1.8.4
#> [13] zlibbioc_1.26.0 munsell_0.4.3 commonmark_1.5 gtable_0.2.0
#> [17] htmlwidgets_1.2 codetools_0.2-15 memoise_1.1.0 evaluate_0.10.1
#> [21] labeling_0.3 knitr_1.20 httpuv_1.4.1 crosstalk_1.0.0
#> [25] Rcpp_0.12.16 xtable_1.8-2 backports_1.1.2 promises_1.0.1
#> [29] scales_0.5.0 desc_1.2.0 jsonlite_1.5 mime_0.5
#> [33] fs_1.2.2 hms_0.4.2 digest_0.6.15 stringi_1.1.7
#> [37] shiny_1.0.5 rprojroot_1.3-2 grid_3.5.0 tools_3.5.0
#> [41] magrittr_1.5 lazyeval_0.2.1 crayon_1.3.4 pkgconfig_2.0.1
#> [45] MASS_7.3-49 xml2_1.2.0 assertthat_0.2.0 rmarkdown_1.9
#> [49] roxygen2_6.0.1 R6_2.2.2 compiler_3.5.0